Comparing different structures¶
Sometimes you want to compare the contact map of two things that have different topologies. This example notebook will run through a couple of common occurences and the options for those.
Looking at a truncated protein¶
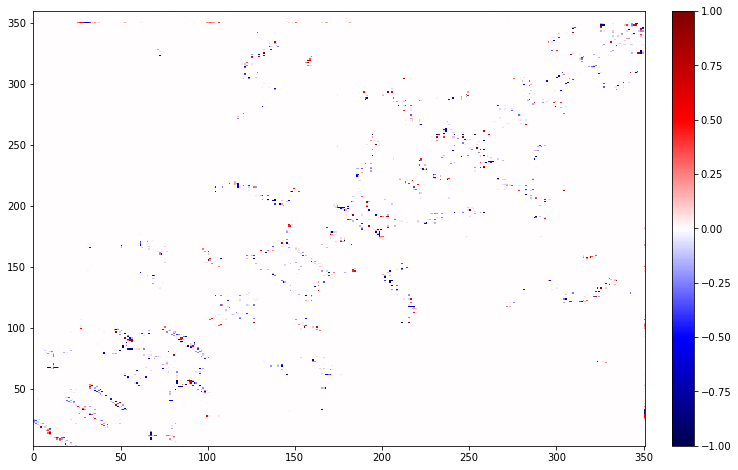
One option is to look at the difference between a truncated protein and the full protein. Here we will fake truncating the protein at residue 150. This works with just subtracting, as that will only care about possible contacts that can be present in both maps.
[1]:
import mdtraj as md
from contact_map import ContactFrequency
[2]:
full = md.load("data/gsk3b_example.h5") # Start with the full trajectory from another example
# Slice another trajectory down to 150 residues
truncated = full.atom_slice(full.topology.select("resid 0 to 150"))
map_full = ContactFrequency(full)
# Here we just use 1 frame so there is a difference to show
map_truncated = ContactFrequency(truncated[0])
[3]:
diff = map_full - map_truncated
# This will only show data for up to residue 150, as differences don't make sense outside of that region
diff.residue_contacts.plot(figsize=(12,8));
Comparing mutated proteins.¶
In order to look at the contact map difference between for example a WT and a mutated protein there are a couple options. We will use the KRAS pdb’s 4OBE (WT) and 5V9O (G12C).
[4]:
WT_full = md.load("data/4obe.pdb") # Load the wildtype structure
# This pdb contains 2 protein chains and we only want one
WT_chain1 = WT_full.atom_slice([i.index for i in WT_full.topology.chain(0).atoms])
G12C_full = md.load("data/5v9o.pdb") # Load the mutated structure
# Cut this one down to just the protein
G12C_protein = G12C_full.atom_slice(G12C_full.topology.select("protein"))
wt_map = ContactFrequency(WT_chain1)
g12c_map = ContactFrequency(G12C_protein)
print(wt_map.topology.residue(11))
print(g12c_map.topology.residue(11))
GLY12
CYS12
If we now try to subtract the two, this will fail because we can’t overlap the atom contact maps
[5]:
#diff = wt_map - g12c_map # This will fail, because the atom indices don't make sense
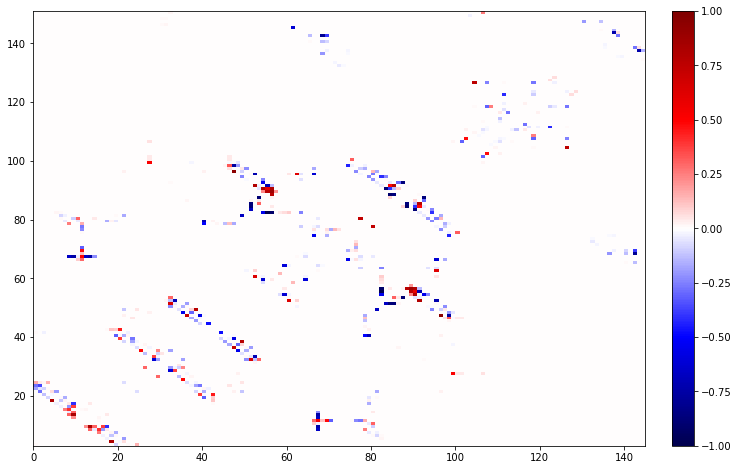
But, we can still look at the residues by using AtomMismatchedContactDifference. All the ContactDifference objects will only show differences between contacts that can be present in the map. So in this case it will not show any differences for residue 168 (GDP) which is only present for the wildtype.
[6]:
from contact_map import AtomMismatchedContactDifference
diff = AtomMismatchedContactDifference(wt_map, g12c_map)
# Grab residue 12
res = diff.topology.residue(11)
# Print the contact differences between WT and G12C (this will print GLY/CYS12 as residue name)
print(diff.residue_contacts.most_common(res))
diff.residue_contacts.plot();
[([GLY/CYS12, LYS16], 0.0)]
Looking at difference when the residue information is missing¶
Sometimes it can happen that the residue information is missing or does not make sense. This is more common for DFT-md simulations than protein simulations, but we will break one of the topologies here to illustrate this behavior and how to handle it
[7]:
full = full
# Make a copy by slicing it to the same number of atoms
broken = full.atom_slice(range(full.topology.n_atoms))
# Here we break the residues, making all resSeqs equal
for i in range(broken.topology.n_residues):
broken.topology.residue(i).resSeq = "broken"
# We also have to break a name, as otherwise mdtraj thinks the topologies are equal
broken.topology.residue(0).name = "test"
map_full = ContactFrequency(full)
map_broken = ContactFrequency(broken[0])
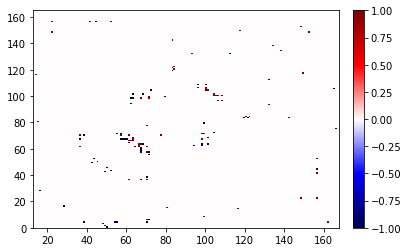
Here the atoms are correct, but we don’t know if the residues would make sense so we can use ResidueMismatchedContactDifference if we only care about the atom-contacts.
[8]:
from contact_map import ResidueMismatchedContactDifference
# diff = map_full - map_broken
diff = ResidueMismatchedContactDifference(map_full, map_broken)
diff.atom_contacts.plot();
/home/sander/github_files/contact_map/contact_map/contact_count.py:238: RuntimeWarning: The number of pixels in the figure is insufficient to show all the contacts.
Please save this as a vector image (such as a PDF) to view the correct result.
Another option is to increase the 'dpi' (currently: 72.0), or the 'figsize' (currently: (6.0, 4.0)).
Recommended minimum amount of pixels = (5641, 5643) (width, height).
warnings.warn(msg, RuntimeWarning)
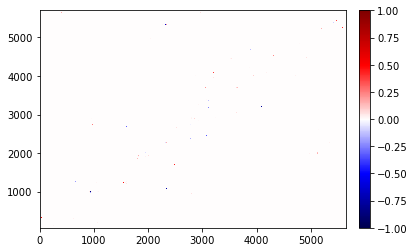
The other option is to override the topology if we have a correct one (in this case full.topology), then we can use OverrideTopologyContactDifference
[9]:
from contact_map import OverrideTopologyContactDifference
diff = OverrideTopologyContactDifference(map_full, map_broken, topology=full.topology)
diff.residue_contacts.plot(figsize=(12,8));